|
|
  |
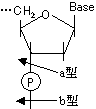 DNaseの場合、一本鎖を切断する酵素と、二本鎖を切断する酵素がある。
DNaseの場合、一本鎖を切断する酵素と、二本鎖を切断する酵素がある。
名称 起源 特異性 被切断鎖 切断様式 膵臓RNase
RNase T1
S1 ヌクレアーゼ
蛇毒ヌクレアーゼ
脾臓ホスホジエステラーゼ
スタフィロコッカスヌクレアーゼ
マングマメヌクレアーゼ
アカパンカビヌクレアーゼ
RNase H
RNase H
膵臓DNase I
Bal 31
Exo I
Exo III
Exo VII
l エキソヌクレアーゼウシ
コウジカビ
A. oryzae
C. atrox
ウシ
S. aureus
マングマメ
アカパンカビ
各種細胞
レトロウィルス
ウシ
A. espejiana
大腸菌
大腸菌
大腸菌
lファージRNA
RNA
RNA,DNA
RNA,DNA
DNA,RNA
RNA,DNA
DNA,RNA
DNA,RNA
RNA,DNA
RNA,DNA
DNA
DNA
DNA
DNA
DNA
DNA
DNA一本鎖
一本鎖
一本鎖
一本鎖
一本鎖
一本鎖
一本鎖
一本鎖
二本鎖
二本鎖
一本鎖,二本鎖
一本鎖
二本鎖
一本鎖
二本鎖
一本鎖
二本鎖エンド(b型)
エンド(b型)
エンド(a型)
エキソ(3'->5')
エキソ(5'->3')
エンド(b型)
エンド(a型)
エンド(a型)
エンド(a型)
エキソ(3'->5')
エンド(a型)
エンド(a型)
エキソ(3'<->5')
エキソ(3'->5')
エキソ(3'->5')
エキソ(3'<->5')
エキソ(5'->3')
|
|
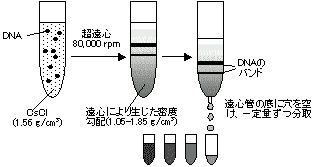
 平衡密度勾配遠心分離 平衡密度勾配遠心分離 |
クロマトグラフィー |
 |
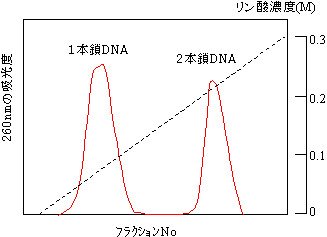 [ヒドロキシアパタイトクロマトグラフィーによる1本鎖と 2本鎖DNAの分離] |
|
 |
||
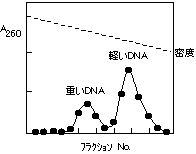
| [CsCl平衡密度勾配遠心法] |
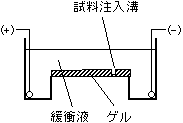
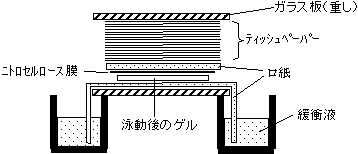
電気泳動
2枚のガラス板の間にアクリル
アミド溶液を入れ固まらせる。
上部にくし型プラスチック板を
入れておくと試料を入れるため
の溝ができる。上下にプラスチックの箱をセット
し、中に泳動用の緩衝液を入
れる。試料を溝に添加し通電。
=>小さな分子ほどゲル
の中を早く移動する[アガロースゲル電気泳動(左)とポリアクリルアミドゲル電気泳動(右)]
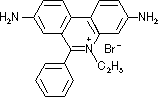
[DNAの染色に用いられる臭化エチジウム(ethidium bromide)]
DNAに結合すると蛍光を出す。
|
|
プロッティング
[ブロッティング装置]
【種々のブロッティング】
(a) サザン(Southern)ブロッティング
標識したmRNAやcDNAでDNA断片を検出する方法
(b) ノザン(Northern)ブロッティング
標識したcDNAdでmRNAを検出する方法
特定のmRNAの(i) 鎖長の解析、(ii) 組織分布、(iii)発現の有無や量の解析に利用される。
(c) ウェスタン(Western)ブロッティング
標識した抗体でタンパク質を検出する方法
★標識法としては、放射能標識以外に蛍光標識がよく利用される。Westernブロッティングでは、酵素標識法や化学発光法も使われる。
ハイブリダイゼーション
[g-32P]ATP
T4 ポリヌクレオチド
キナーゼ[DNAの5'末端の放射性標識法] 広範囲に標識するときはDNAポリメラーゼ I のニックトランスレーションを利用。
[ハイブリダイゼーション]
|
|
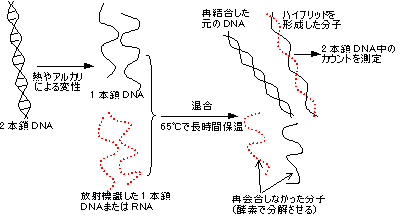
化学的分解法(Maxam-Gilbert法)
- (例)次の13残基のオリゴヌクレオチドの構造決定を行うこととする。
- 5' ATCGATTCTCGGA 3'
- 1) まず、このオリゴヌクレオチドの5'末端を32Pや33Pで放射標識する(●)。
- 5' ●-ATCGATTCTCGGA 3'
- 2) この試料溶液を4等分し、次の4種の化学分解を行う。
切断される塩基 G G + A C C + T 反応液の組成 32P-DNA
DMA緩衝液
ジメチル硫酸32P-DNA
水
ギ酸32P-DNA
NaCl溶液
ヒドラジン32P-DNA
水
ヒドラジン反応条件 20℃,4.5分 20℃,10分 20℃,8分 20℃,8分
- この条件では、特定の塩基のところでランダムに切断が起こり、反応液中に以下のような放射標識断片が生じる。(標識されていない断片もたくさんできるが,検出できないので考えなくてよい)
Gで分解 Cで分解 ●-ATC
●-ATCGATTCTC
●-ATCGATTCTCG●-AT
●-ATCGATT
●-ATCGATTCTG + Aで分解 C + T で分解 ●
●-ATC
●-ATCG
●-ATCGATTCTC
●-ATCGATTCTCG
●-ATCGATTCTCGG
●-A
●-AT
●-ATCGA
●-ATCGAT
●-ATCGATT
●-ATCGATTC
●-ATCGATTCT
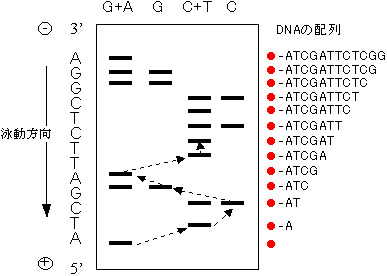
- 3) これら4つの試料をポリアクリルアミドゲル電気泳動にかける。各々のDNA断片はサイズだけで分離される。
4) ゲルのオートラジオグラムを作成する(X線フィルムに露光すると、例の場合、図のようになるであろう。
[Maxam-Gilbert法によるDNA塩基配列の決定]
(オートラジオグラム)
- 5) 図の泳動先端(図の下端)のバンドから順に上にバンドの位置をたどっていくことで、目的DNA断片の5'→3'方向の配列を決定できる。(ただし、C+TとCの同じ位置にバンドがある場合はCとなり、C+TにはあるがCにバンドがない場合はTとなる。G+Aについても同様。)
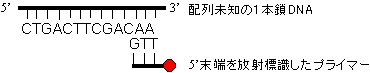
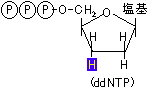
ジデオキシ法(Sanger*法, 鎖終結法)1) まず、配列を決めたい1本鎖DNAの3'末端に相補的な短いオリゴヌクレオチドをプライマーとして準備し、プライマーの5'末端を33Pや35Sで放射標識するか,または蛍光色素で標識する。
5' CTGACTTCGACAA 3' 未知の塩基配列をもつ1本鎖DNA 3' GTT●
5' 5'標識プライマー
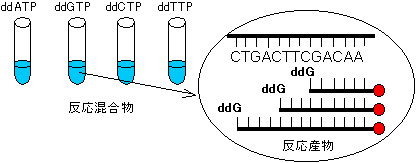
2) 配列を決めたい1本鎖DNA溶液を4等分し、標識プライマー、4種のdNTPを加える。これらにterminator(ddNTP)を1種ずつ加える。
3) DNAポリメラーゼIのKlenowフラグメントを加えて複製を開始する。
->加えたddNTPのため、鎖伸長はランダムな位置で停止する。
DNAポリメラーゼI
dATP
dGTP
dCTP
dTTP例えばddATPを少量加えた試験管の場合、上の配列では次の
3つの標識断片が生成する。下線はプライマー部分を表す。4) これら4つの試料をポリアクリルアミドゲル電気泳動にかける。各々のDNA断片はサイズだけで分離される。
ddAGCTGTT-● 5'
ddAAGCTGTT-● 5'
ddACTGAAGCTGTT-● 5'
5) ゲルのオートラジオグラムを作成する。
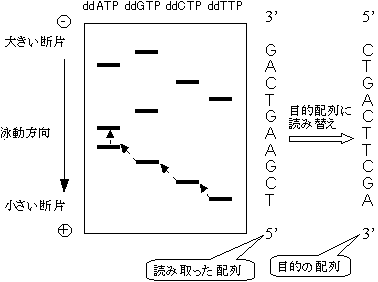
6) 図のように、泳動先端(下端)のバンドから順に上にバンドの位置を辿っていくことで、目的配列の相補鎖の5'→3'方向の配列を決定できる。
[Sanger法による塩基配列決定]
これを目的配列(鋳型鎖)に読み替えるには、読みとった配列をひっくり返せばよい。
蛍光標識法を用いる塩基配列決定の自動化
- Sangerの原法では、実験ごとにプライマーをいちいち放射標識する必要があり、大変面倒である。波長の異なる蛍光発色団でterminatorを標識することにより、迅速で簡便な塩基配列決定法が開発された。
ddATP-● ddTTP-●
ddGTP-● ddCTP-●
波長の異なる4種の蛍光発色団
実際には、rhodamine系蛍光色素[蛍光標識terminators]
≪蛍光標識法の手順≫
- 先ず、配列を決めたい1本鎖DNA、プライマー、4種のdNTPを含む溶液に、波長の異なる蛍光色素で標識した4種のddNTP混合物(terminator)を加える。
- DNAポリメラーゼで鎖を伸長させる。加えたterminatorによって、鎖伸長はランダムな位置で停止する。
- 試料をゲル電気泳動にかける。
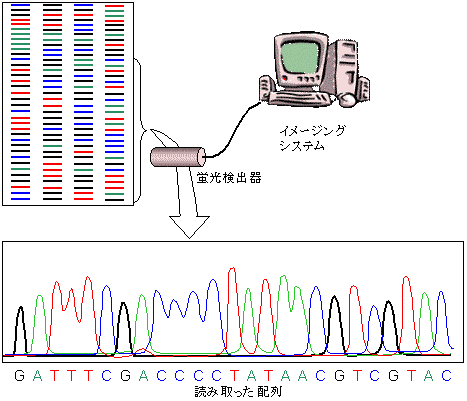
- ゲルにレーザー光を当てて蛍光バンドを検出し、情報をイメージングシステムに転送する(図 8.11)。
- 検出したバンドの情報をコンピューターで処理し、ピークとして表す。
[蛍光標識法による塩基配列決定]
| 《蛍光標識法の利点》 1. プライマーを標識する必要がない。 2. 反応は1本の試験管で行える。 3. 電気泳動も1試料1レーンですむため、バンドの物理的なゆがみによる読み取り誤差がない。 また、大量の試料を一度に処理できる。 4. 放射能を使用しないので、いつでも実施でき、また、放射性廃棄物を出さない。 5. オートラジオグラフィーが不要。 6. 自動化ができるので、迅速で簡便。 |
|
|
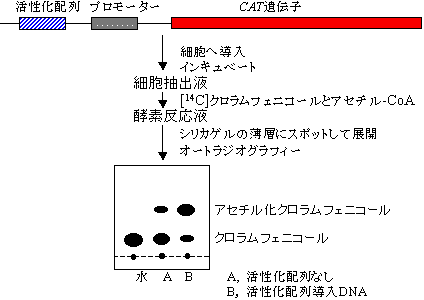
転写活性の測定法
転写制御因子などのDNA結合タンパク質の検出
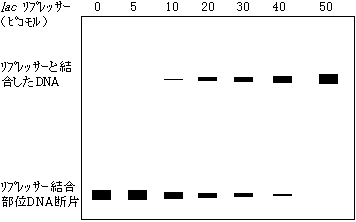
- 1) ゲルシフト法(電気泳動移動度シフト解析: electrophoretic mobility shift assay, EMSA)
- タンパク質(転写制御因子)が結合すると、分子サイズが大きくなり電気泳動におけるDNAの移動度が低下することを利用した転写制御因子の検出法。32Pで標識したDNA断片と転写制御因子を混ぜ、ゲル電気泳動にかける。オートラジオグラフィーでDNAの位置を見ると、因子の結合したDNAはゆっくり動くので、通常のバンドよりも遅れて移動するバンドとして検出される。DNAのほんの一部に結合があっても検出でき、DNA結合反応の定量的解析ができる。
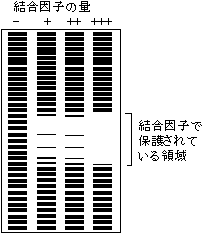
- 2) DNase I フットプリント法
- DNA分解酵素の一種であるDNase I (デオキシリボヌクレアーゼⅠ)は裸のDNAを分解するが、タンパク質と結合したDNAは分解しない。この性質を利用して、転写制御因子が結合するDNAの領域を決定する方法である。一端を32Pで標識したDNA断片と転写制御因子を混ぜ、少量のDNase Iを低温または短時間作用させて部分的にDNAを分解する。電気泳動とオートラジオグラフィーでDNAの断片を調べる。標識末端からのDNA長に従ってはしご状のバンドが見えるが、転写制御因子が結合した部分ではDNAが切れないために断片が生じず、はしご状のバンドの一部が足跡(footprint)のように抜ける。同じDNA断片をMaxam-Gilbert法で処理したものと比較することで、抜け落ちた領域が決定できる。
in situ ハイブリッド形成法(in situ hybridization)
 |
加熱処理 変性 |
 |
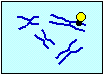
ビオチン化した DNAプローブ ハイブリダイズ |
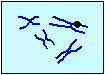 |
蛍光標識した アビジン ビオチン化DNA が結合した部位を |
| スライドガラス上の 二本鎖DNA |
一本鎖DNA | 標的配列に結合した プローブ(●) |
視覚化する | ||
 |
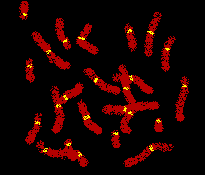 ヒト染色体セントロメアのサテライト DNA ビオチン標識したDNAプローブの 結合した位置に蛍光標識アビジンが 結合し、赤く対染色した染色体を 背景に,黄色の蛍光で観察できる。 |
||||
| ビオチンーアビジンを 介して蛍光色素(●) が標的DNAに結合 |
|||||
遺伝子歩行(gene walking)
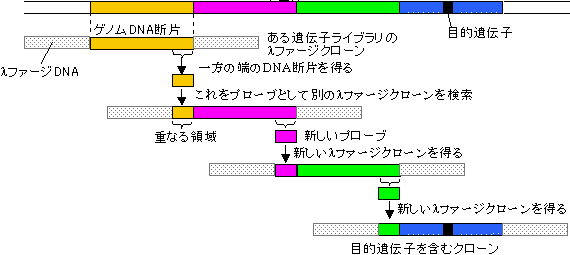
| 《一言》 ヒト血液凝固因子VIII遺伝子は190kbp、Duchenne筋ジストロフィーの原因タンパク質(ジストロフィン)遺伝子は2,000kbpにもわたる。また、真核細胞の遺伝子間の距離は一般に非常に遠いため、遺伝子歩行のためにはできるだけ大きなクローンが有利である。 =>YACベクター |
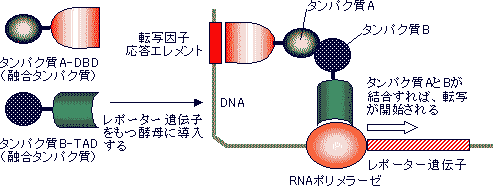
ツーハイブリッド法
|
|